Каждый год Институт биоинформатики в Санкт-Петербурге и Москве набирает биологов, математиков и программистов, чтобы погрузить в мир биоинформатики. Биологи учатся программировать и тренируются реализовывать идеи в коде, а информатики изучают биологию и применяют алгоритмические подходы к биологическим и медицинским задачам. Самая важная часть обучения — реальные научные проекты. В этой статье мы расскажем о работе и результатах студентов Института, сделанной под руководством Олега Шпынова из JetBrains Research в 2019 году. Проект посвящен изучению изменения хроматина человека с помощью машинного обучения.

Студенты-информатики 2019 Института биоинформатики
Желание удовлетворить любопытство и понять себя, которое началось с описания анатомии человека, постепенно углублялись и переходили на более детальный уровень. Изучались клетки крови и их взаимодействие с паразитами, механизмы передачи наследственной информации и образования метастаз раковыми клетками.
Появление технологий секвенирования позволило перейти на еще один уровень глубже и смотреть непосредственно «в лицо» носителю генетической информации — ДНК. Иначе говоря, дезоксирибонуклеиновой кислоте, которая находится в ядре почти каждой клетки нашего организма и отвечает за то, как мы выглядим, какого роста, каким тембром голоса говорим и можем ли заболеть малярией. Однако технологии, как и биохимические методы, не стоят на месте. Их комбинация позволила «выводить на свет» более сложные механизмы работы организма. Давайте разберемся с этим подробнее.
Технологии секвенирования изменялись, и теперь технологический прогресс позволяет в зависимости от пожеланий секвенировать отдельно клетки, смотреть изменения в них во времени или просто получить полную информацию о последовательности носителя наследственной информации — ДНК. По сути, секвенирование позволяет переводить биологическую молекулу в текстовый файл, с которым потом можно работать как с обычным текстом. Современные методы секвенирования используют подход «дробовика»и дают на выходе огромное количество коротких фрагментов. В некоторых анализах эти короткие фрагменты «примеряют» на уже существующих геном и смотрят различия в последовательности «текста».
Нить ДНК очень длинная и постоянно находиться в раскрученном состоянии не может — это неудобно и опасно (больше вероятность того, что где-то будет разрыв). Поэтому молекула спирализуется (сильно-сильно скручивается) и компактно упаковывается, накручиваясь на специальные белковые комплексы, как волосы на бигуди. Эти комплексы зовутся нуклеосомами и состоят из белков гистонов. Модификация гистонов является одним из примеров более общего механизма эпигенетической регуляции. Организм живой и ему необходимо реагировать на окружающие изменения. Реакция организма заключается в том числе и в изменении экспрессии генов. Если фрагмент ДНК, на которой расположен ген, плотно упакован и намотан на нуклеосому, то подобраться к нему и считать информацию невозможно. Поэтому на гистоны навешиваются особые фосфорильные и ацетильные группы, происходит так называемое фосфорилирование или ацетилирование. Это заставляет гистон «подвинуться» и дать доступ к нужному фрагменту ДНК. Но нуклеосома все равно остается связанной с ДНК и это можно использовать при исследовании регуляции.
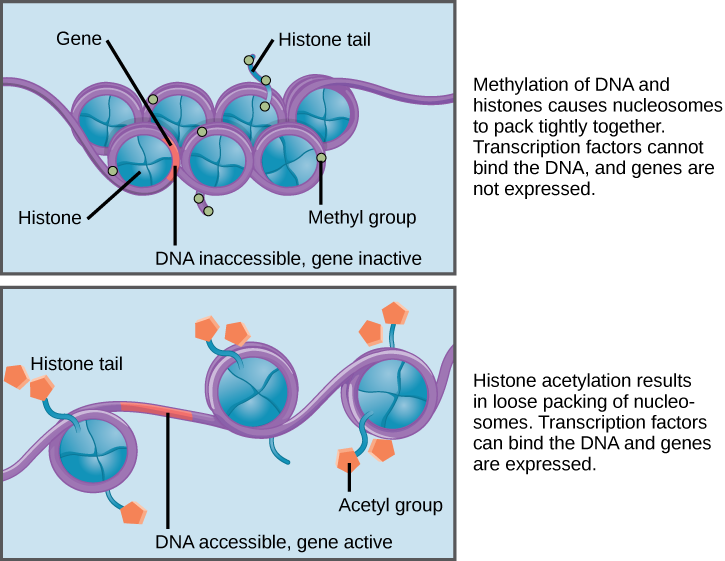
Механизм ацетилирования и метилирования гистонов (источник)
Для изучения фрагментов ДНК, которые остаются связаны с белком, существует специальный метод: иммунопреципитация хроматина (chromatin immunoprecipitation, ChIP). Происходит этот анализ следующим образом:
Если говорить коротко, мы вытаскиваем из раствора белок, сцепленный с ДНК, и заставляем его «отпустить» ДНК. С биологической точки зрения поле действия понятно: изучение экспрессии генов, закрытых и открытых областей и т.д. О вещах, которые могут делать в этой задаче программисты, мы расскажем ниже.
В случае ChIP секвенирования (-seq) получившиеся фрагменты ДНК подвергаются амплификации (искусственное дублирование фрагментов) и секвенированию. Набор последовательностей маленьких кусочков ДНК и изучают биоинформатики.
Полученные данные проходят контроль качества, фильтруются, выравниваются на последовательность ДНК и обрабатываются специальными программами.

Схема получения ДНК для анализа
Задача поиска мест связывания на ДНК часто называется задачей поиска пиков (peak calling), и класс инструментов — peak callers. На данный момент существует множество вычислительных подходов и инструментов для анализа таких данных, однако, алгоритмы не идеальны и имеют целый ряд ограничений. В этой области все еще остается много нерешенных вычислительных задач для программистов и информатиков.
Вот некоторые из них, которые в настоящее время решают студенты математических и технических специальностей:
Доступность хроматина при фрагментации неодинакова в разных частях генома: в активно транскрибируемых областях он доступнее, поэтому соответствующие фрагменты ДНК будут преобладать в образце, что может привести к ложноположительному результату. Плотно упакованные участки, напротив, могут хуже подвергаться фрагментации и, следовательно, будут менее представлены в образце, что может привести к ложноотрицательному результату
У классической методики существует ряд ограничений. Так, обычно для ChIP-seq необходимо значительное количество клеток (около 10 миллионов), что затрудняет применение данного метода на маленьких организмах (типа грибов или простейших), а также ограничивает количество экспериментов, которые можно провести с ценным образцом.
В ходе проведения эксперимента ChIP-seq существует вероятность получить в итоговой библиотеке не только фрагменты ДНК, которые были связаны с белком, но и другие, неспецифично связанные фрагменты. Это может происходить вследствие не идеальной специфичности антитела, проблемы с отмывкой свободных фрагментов ДНК, и т.д. Такие фрагменты и образуют так называемый шум в данных. Проблема заключается не только в существовании шума, но и в сложности его измерения. Для оценки его уровня существует метрика отношение сигнал/шум (SNR, signal-to-noise ratio), которая определяется числом и мощностью пиков, полученных для каждого образца. Однако высокое значение SNR не гарантирует правильность определения сайтов связывания, а всего лишь отражает наличие большого количества участков генома, на которые выровнялось (на хромосоме в данном месте последовательность совпадает с искомой) много ридов — маленьких фрагментов ДНК.
Часть этих задач решали студенты Института биоинформатики под руководством Олега Шпынова из JetBrains Research в рамках семестровых научных проектов.
Noisy peak calling.
студентка: Чаплыгина Дарья

В статье “Impact of sequencing depth in ChIP-seq experiments” (1) авторы изучили влияние размера библиотеки (количества исходных ридов) на результаты работы алгоритмов поиска пиков. Они создали искусственные наборы данных для разных типов гистоновых модификации путем случайного сэмплирования из реальных экспериментов. Как и ожидалось, чем беднее библиотека, тем сложнее алгоритмам находить пики, результаты получаются несогласованными между разными методами. Но еще они заметили, что, в случае использования одного и того же инструмента, теряется согласованность и между биологическими повторностями. В семестровом проекте мы исследовали влияние уровня шума в исходных данных.
Набор данных с контролируемым уровнем шума был получен на основе находящихся в открытом доступе данных ChIP-seq экспериментов с сайта ENCODE project ENCODE project. Для этого было использовано две модели шума:
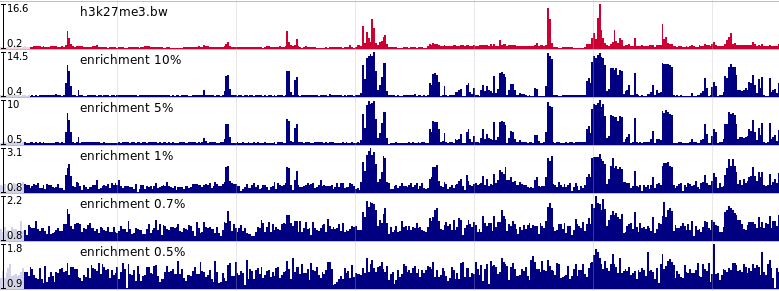
Визуализация изменений в данных при применении вероятностной модели шума
На полученном наборе данных мы проанализировали три алгоритма: MACS2 (2), SICER (3) и SPAN (алгоритм, разрабатываемый JetBrains Research. В основе лежит semi-supervised метод машинного обучения). Как оказалось, при фиксированном SNR можно предсказать ожидаемую точность и полноту множества пиков, которое будет найдено алгоритмом. При высоком уровне шума (или низком SNR): MACS2 и SICER почти не находят пиков, SPAN же демонстрирует наиболее стабильные результаты по совокупности показателей.

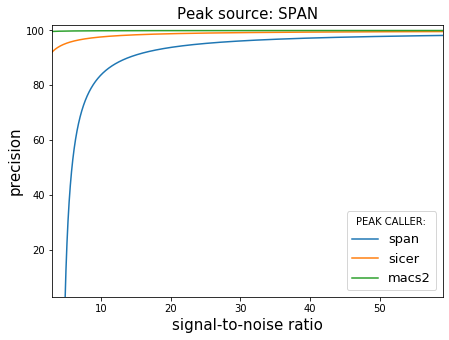
Точность и полнота работы алгоритмов поиска пиков в условиях контролируемого уровня шума
Мы изучили, как в процессе зашумления меняются две метрики качества данных: SNR и процент фрагментов внутри пиков (FRIP — Fraction of Reads In Peaks). Измерения показали, что при одинаковом значении SNR доля фрагментов, приходящихся на регионы взаимодействия ДНК с белком может значительно варьироваться (в некоторых случаях разница составляла до 50%). Существующие стандарты и рекомендации по оценке качества данных экспериментов ChIP-seq неполны, требуется создание новых комплексных подходов.
В рамках работы мы также разработали пайплайны для полу-автоматического проведения подобных экспериментов.
Реализация подходов и исходный код:
github.com/DaryaChaplygina/NoisyPeakCalling,
github.com/DaryaChaplygina/NoisyPeakCalling2.
Deep learning to the rescue!
студентка: Дарья Балашова
Одним из ограничений классического метода ChIP-seq является большое количество необходимого клеточного материала, что не позволяет провести эксперимент, например, в случае редких клеточных популяций или в случае нескольких измерений для одного биологического образца. Новый метод Ultra-Low-Input (ULI) ChIP-seq (4) требует значительно меньше материала — достаточно 100 тысяч клеток — но имеет большую вариативность и уровень шума в данных.
Использование методов глубокого машинного обучение набирает популяность в биоинформатике, демонстрируя отличные результаты в решении таких задач, как обработка биомедицинских изображений. В работе “Denoising genome-wide histone ChIP-seq with convolutional neural networks” (5) авторы предложили алгоритм Coda — метод улучшения качества данных ChIP-seq на основе сверточных нейронных сетей (Convolutional Neural Network). Они создали и обучили глубокую нейронную сеть не только улучшать данные плохого качества, но и находить в них пики.
В рамках данного проекта оригинальный алгоритм был адаптирован для ULI ChIP-seq данных. Используя наработки из предыдущего проекта и данные ULI ChIP-seq из статьи “Epigenetic changes in aging human monocytes” (6) мы проанализировали такие важные характеристики работы алгоритма, как улучшение метрик качества, например, SNR. В результате был создан алгоритм DCNN — сверточная нейронная сеть для автоматического улучшения качества в данных на основе соотношения сигнал/шум в случае наличия биологических повторностей. Если улучшения и очистка сигнала работает довольно хорошо, то поиск мест связывания белков с ДНК при помощи методов глубоко обучения все еще является нерешенной проблемой, так как существующие подходы предполагают наличие большой и качественной обучающей выборки.
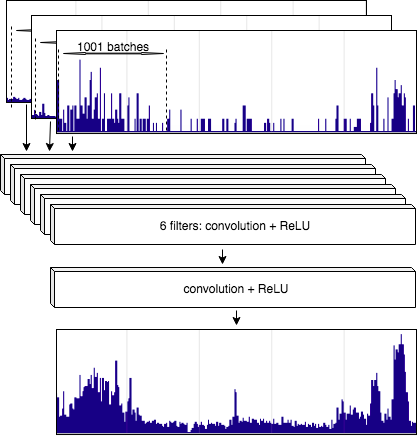
Схематическое изображение применения сверточной нейронной сети DCNN
Реализация подхода и исходный код: github.com/dashabalashova/Denoising_CNN.
Биоинформатика позволяет применить подходы программистов к биологическим данным и получать новые знания, которые помогут биологам и медикам изучать человека. Сейчас открыт приём заявок на летнюю школу 2020, которая пройдёт в Санкт-Петербурге с 27 июля по 1 августа. Она идеально подойдет для знакомства с биоинформатикой.
Для тех, кто решился на более серьезное обучение — есть шанс вскочить в последний вагон и подать заявку до 22 февраля на программу переподготовки по биоинформатике в Петербурге и Москве или до 1 марта нa выездной семинар по системной биологии.
Для любителей читать и открывать новое у нас есть список книг и учебников по алгоритмам, программированию, генетике и биологии.
Авторы статьи:
Ольга Бондарева, Институт биоинформатики
Олег Шпынов, JetBrains Research
Екатерина Вяххи, Институт биоинформатики

Студенты-информатики 2019 Института биоинформатики
Что такое секвенирование и зачем оно нужно
Желание удовлетворить любопытство и понять себя, которое началось с описания анатомии человека, постепенно углублялись и переходили на более детальный уровень. Изучались клетки крови и их взаимодействие с паразитами, механизмы передачи наследственной информации и образования метастаз раковыми клетками.
Появление технологий секвенирования позволило перейти на еще один уровень глубже и смотреть непосредственно «в лицо» носителю генетической информации — ДНК. Иначе говоря, дезоксирибонуклеиновой кислоте, которая находится в ядре почти каждой клетки нашего организма и отвечает за то, как мы выглядим, какого роста, каким тембром голоса говорим и можем ли заболеть малярией. Однако технологии, как и биохимические методы, не стоят на месте. Их комбинация позволила «выводить на свет» более сложные механизмы работы организма. Давайте разберемся с этим подробнее.
Как мы секвенируем организмы
Технологии секвенирования изменялись, и теперь технологический прогресс позволяет в зависимости от пожеланий секвенировать отдельно клетки, смотреть изменения в них во времени или просто получить полную информацию о последовательности носителя наследственной информации — ДНК. По сути, секвенирование позволяет переводить биологическую молекулу в текстовый файл, с которым потом можно работать как с обычным текстом. Современные методы секвенирования используют подход «дробовика»и дают на выходе огромное количество коротких фрагментов. В некоторых анализах эти короткие фрагменты «примеряют» на уже существующих геном и смотрят различия в последовательности «текста».
Что такое гистоны и на что они влияют
Нить ДНК очень длинная и постоянно находиться в раскрученном состоянии не может — это неудобно и опасно (больше вероятность того, что где-то будет разрыв). Поэтому молекула спирализуется (сильно-сильно скручивается) и компактно упаковывается, накручиваясь на специальные белковые комплексы, как волосы на бигуди. Эти комплексы зовутся нуклеосомами и состоят из белков гистонов. Модификация гистонов является одним из примеров более общего механизма эпигенетической регуляции. Организм живой и ему необходимо реагировать на окружающие изменения. Реакция организма заключается в том числе и в изменении экспрессии генов. Если фрагмент ДНК, на которой расположен ген, плотно упакован и намотан на нуклеосому, то подобраться к нему и считать информацию невозможно. Поэтому на гистоны навешиваются особые фосфорильные и ацетильные группы, происходит так называемое фосфорилирование или ацетилирование. Это заставляет гистон «подвинуться» и дать доступ к нужному фрагменту ДНК. Но нуклеосома все равно остается связанной с ДНК и это можно использовать при исследовании регуляции.

Механизм ацетилирования и метилирования гистонов (источник)
Хроматин-иммунопреципитационное секвенирование (ChIP-seq) и его применение
Для изучения фрагментов ДНК, которые остаются связаны с белком, существует специальный метод: иммунопреципитация хроматина (chromatin immunoprecipitation, ChIP). Происходит этот анализ следующим образом:
- образование обратимых сшивок между ДНК и взаимодействующими с ней белками (обычно с помощью обработки формальдегидом)
- выделение ДНК и расщепление на фрагменты ультразвуком или эндонуклеазами
- осаждение специфическими к исследуемому белку антителами
- разрушение сшивок между белком и ДНК, очистка ДНК
Если говорить коротко, мы вытаскиваем из раствора белок, сцепленный с ДНК, и заставляем его «отпустить» ДНК. С биологической точки зрения поле действия понятно: изучение экспрессии генов, закрытых и открытых областей и т.д. О вещах, которые могут делать в этой задаче программисты, мы расскажем ниже.
В случае ChIP секвенирования (-seq) получившиеся фрагменты ДНК подвергаются амплификации (искусственное дублирование фрагментов) и секвенированию. Набор последовательностей маленьких кусочков ДНК и изучают биоинформатики.
Полученные данные проходят контроль качества, фильтруются, выравниваются на последовательность ДНК и обрабатываются специальными программами.

Схема получения ДНК для анализа
Задача поиска мест связывания на ДНК часто называется задачей поиска пиков (peak calling), и класс инструментов — peak callers. На данный момент существует множество вычислительных подходов и инструментов для анализа таких данных, однако, алгоритмы не идеальны и имеют целый ряд ограничений. В этой области все еще остается много нерешенных вычислительных задач для программистов и информатиков.
Вот некоторые из них, которые в настоящее время решают студенты математических и технических специальностей:
- Неравномерная фрагментация и контроль
Доступность хроматина при фрагментации неодинакова в разных частях генома: в активно транскрибируемых областях он доступнее, поэтому соответствующие фрагменты ДНК будут преобладать в образце, что может привести к ложноположительному результату. Плотно упакованные участки, напротив, могут хуже подвергаться фрагментации и, следовательно, будут менее представлены в образце, что может привести к ложноотрицательному результату
- Количество клеток
У классической методики существует ряд ограничений. Так, обычно для ChIP-seq необходимо значительное количество клеток (около 10 миллионов), что затрудняет применение данного метода на маленьких организмах (типа грибов или простейших), а также ограничивает количество экспериментов, которые можно провести с ценным образцом.
- Шум в данных
В ходе проведения эксперимента ChIP-seq существует вероятность получить в итоговой библиотеке не только фрагменты ДНК, которые были связаны с белком, но и другие, неспецифично связанные фрагменты. Это может происходить вследствие не идеальной специфичности антитела, проблемы с отмывкой свободных фрагментов ДНК, и т.д. Такие фрагменты и образуют так называемый шум в данных. Проблема заключается не только в существовании шума, но и в сложности его измерения. Для оценки его уровня существует метрика отношение сигнал/шум (SNR, signal-to-noise ratio), которая определяется числом и мощностью пиков, полученных для каждого образца. Однако высокое значение SNR не гарантирует правильность определения сайтов связывания, а всего лишь отражает наличие большого количества участков генома, на которые выровнялось (на хромосоме в данном месте последовательность совпадает с искомой) много ридов — маленьких фрагментов ДНК.
Варианты решения проблем
Часть этих задач решали студенты Института биоинформатики под руководством Олега Шпынова из JetBrains Research в рамках семестровых научных проектов.
Noisy peak calling.
студентка: Чаплыгина Дарья

В статье “Impact of sequencing depth in ChIP-seq experiments” (1) авторы изучили влияние размера библиотеки (количества исходных ридов) на результаты работы алгоритмов поиска пиков. Они создали искусственные наборы данных для разных типов гистоновых модификации путем случайного сэмплирования из реальных экспериментов. Как и ожидалось, чем беднее библиотека, тем сложнее алгоритмам находить пики, результаты получаются несогласованными между разными методами. Но еще они заметили, что, в случае использования одного и того же инструмента, теряется согласованность и между биологическими повторностями. В семестровом проекте мы исследовали влияние уровня шума в исходных данных.
Набор данных с контролируемым уровнем шума был получен на основе находящихся в открытом доступе данных ChIP-seq экспериментов с сайта ENCODE project ENCODE project. Для этого было использовано две модели шума:
- Аддитивная модель. В исходный файл с «чистыми данными» добавлялись фрагменты со случайных участков ДНК. Доля случайных фрагментов варьировалась от 0% до 90%.
- Вероятностная модель. Для каждого эксперимента строилась математическая модель с использованием инструмента Tulip. С ее помощью генерировался полностью новый эксперимент, один из параметров которого – процент фрагментов, которые расположены внутри сайтов связывания ДНК с белком, – менялся от 10% до 0.5%.

Визуализация изменений в данных при применении вероятностной модели шума
На полученном наборе данных мы проанализировали три алгоритма: MACS2 (2), SICER (3) и SPAN (алгоритм, разрабатываемый JetBrains Research. В основе лежит semi-supervised метод машинного обучения). Как оказалось, при фиксированном SNR можно предсказать ожидаемую точность и полноту множества пиков, которое будет найдено алгоритмом. При высоком уровне шума (или низком SNR): MACS2 и SICER почти не находят пиков, SPAN же демонстрирует наиболее стабильные результаты по совокупности показателей.


Точность и полнота работы алгоритмов поиска пиков в условиях контролируемого уровня шума
Мы изучили, как в процессе зашумления меняются две метрики качества данных: SNR и процент фрагментов внутри пиков (FRIP — Fraction of Reads In Peaks). Измерения показали, что при одинаковом значении SNR доля фрагментов, приходящихся на регионы взаимодействия ДНК с белком может значительно варьироваться (в некоторых случаях разница составляла до 50%). Существующие стандарты и рекомендации по оценке качества данных экспериментов ChIP-seq неполны, требуется создание новых комплексных подходов.
В рамках работы мы также разработали пайплайны для полу-автоматического проведения подобных экспериментов.
Реализация подходов и исходный код:
github.com/DaryaChaplygina/NoisyPeakCalling,
github.com/DaryaChaplygina/NoisyPeakCalling2.
Deep learning to the rescue!
студентка: Дарья Балашова
Одним из ограничений классического метода ChIP-seq является большое количество необходимого клеточного материала, что не позволяет провести эксперимент, например, в случае редких клеточных популяций или в случае нескольких измерений для одного биологического образца. Новый метод Ultra-Low-Input (ULI) ChIP-seq (4) требует значительно меньше материала — достаточно 100 тысяч клеток — но имеет большую вариативность и уровень шума в данных.
Использование методов глубокого машинного обучение набирает популяность в биоинформатике, демонстрируя отличные результаты в решении таких задач, как обработка биомедицинских изображений. В работе “Denoising genome-wide histone ChIP-seq with convolutional neural networks” (5) авторы предложили алгоритм Coda — метод улучшения качества данных ChIP-seq на основе сверточных нейронных сетей (Convolutional Neural Network). Они создали и обучили глубокую нейронную сеть не только улучшать данные плохого качества, но и находить в них пики.
В рамках данного проекта оригинальный алгоритм был адаптирован для ULI ChIP-seq данных. Используя наработки из предыдущего проекта и данные ULI ChIP-seq из статьи “Epigenetic changes in aging human monocytes” (6) мы проанализировали такие важные характеристики работы алгоритма, как улучшение метрик качества, например, SNR. В результате был создан алгоритм DCNN — сверточная нейронная сеть для автоматического улучшения качества в данных на основе соотношения сигнал/шум в случае наличия биологических повторностей. Если улучшения и очистка сигнала работает довольно хорошо, то поиск мест связывания белков с ДНК при помощи методов глубоко обучения все еще является нерешенной проблемой, так как существующие подходы предполагают наличие большой и качественной обучающей выборки.

Схематическое изображение применения сверточной нейронной сети DCNN
Реализация подхода и исходный код: github.com/dashabalashova/Denoising_CNN.
Вместо послесловия
Биоинформатика позволяет применить подходы программистов к биологическим данным и получать новые знания, которые помогут биологам и медикам изучать человека. Сейчас открыт приём заявок на летнюю школу 2020, которая пройдёт в Санкт-Петербурге с 27 июля по 1 августа. Она идеально подойдет для знакомства с биоинформатикой.
Для тех, кто решился на более серьезное обучение — есть шанс вскочить в последний вагон и подать заявку до 22 февраля на программу переподготовки по биоинформатике в Петербурге и Москве или до 1 марта нa выездной семинар по системной биологии.
Для любителей читать и открывать новое у нас есть список книг и учебников по алгоритмам, программированию, генетике и биологии.
Список литературы:
- Jung, Y. L., Luquette, L. J., Ho, J. W., Ferrari, F., Tolstorukov, M., Minoda, A.,… & Park, P. J. (2014). Impact of sequencing depth in ChIP-seq experiments. Nucleic acids research, 42(9), e74-e74.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E.,… & Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome biology, 9(9), R137.
- Xu, S., Grullon, S., Ge, K., & Peng, W. (2014). Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. In Stem Cell Transcriptional Networks (pp. 97-111). Humana Press, New York, NY.
- Brind’Amour, J., Liu, S., Hudson, M., Chen, C., Karimi, M. M., & Lorincz, M. C. (2015). An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nature communications, 6(1), 1-8.
- Koh, P. W., Pierson, E., & Kundaje, A. (2017). Denoising genome-wide histone ChIP-seq with convolutional neural networks. Bioinformatics, 33(14), i225-i233.
- Schukina, Bagaitkar, Shpynov et al., в ревью, сайт artyomovlab.wustl.edu/aging
Авторы статьи:
Ольга Бондарева, Институт биоинформатики
Олег Шпынов, JetBrains Research
Екатерина Вяххи, Институт биоинформатики
