Еще летом я запланировал эксперимент и написал статью Использование UML для эксперимента по эволюционной систематике прокариот, и косвенно о психологии ученых. Результаты по грубой обработки уже были готовы к концу лета (спасибо, mktums за помощь ).
Вот теперь образовалась пауза, и я добил эту тему, и представляю результаты.
(кое-что повторю из предыдущей статьи, чтобы не заставлять новых читателей ее читать)
Основная критика статьи Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение» заключалась в следующей претензии "Нельзя рассматривать один ген в качестве мерила". С этим я полностью согласен, и этот эксперимент это исправляет в полной мере.
Немного цифр. Сейчас в NCBI есть порядка 2000 геномов бактерий (3723 локусов). При подготовке к эксперименту, я выделил все тРНК, которые помечены таким образом. Их оказалось более 40 тысяч уникальных вариаций. Но увы, среди них много ошибок (порядка 50%, см. предыдущие статьи, где это обсуждалось подробно).
Но я подумал, что можно пропустить этап полноценного исправления ошибок. Как это сделать? Я рассортировал указанные тРНК по длине и по наличию конца CCA на конце последовательности. Надо сказать, что последовательность CCA обязательна для любой тРНК, а длина может быть от 74 до 96 нуклеотидов.
В NCBI есть много чудес вплоть до тРНК из одного нуклеотида, или более 1300 :) (без улыбки не скажешь). Поэтому я убрал последовательности, которые имеют длину до 70 и больше 100, а также те которые не оканчиваются на CCA.
Их стало около 20000. Это наиболее вероятные тРНК, которые не содержат ошибок из NCBI. С оставшийся половиной тРНК — можно разобраться позже.
На самом деле, для планируемого эксперимента без разницы содержит ли ошибки данная конкретная последовательность длиной 70-100 нуклеотидов или нет. Почему? Так как я собираюсь перепроверить по геномам 2000 бактерий, действительно ли есть такие последовательности — ошибки будут исключены. А тРНК это на самом деле или нет это дело второе. Главное, что у разных организмов совпадают значительные участки ДНК. Совпадение последовательности длиной 70-100 в геномах — дело далеко не случайное.
Поэтому, что теперь я делаю. Беру этих 20000 тРНК и нахожу в каких бактериях они присутствуют. Если последовательность присутствует только в одном организме — это не интересно. И скорее всего это ошибочная последовательность. И таким образом отсеивается еще солидный процент ошибок.
Если же последовательность есть в более чем в одном организме — это одна ассоциация (связь) между двумя организмами.
В первой статье был сделан важный вывод, что
Также мне посоветовали отобразить граф с помощью Graphviz, что я и сделал. Но Graphviz зависает, когда число связей в графе больше 1000. А общий граф у меня получился на 6172 связи. Поэтому тут показываю лишь маленький фрагмент для наглядности. И даю ссылку на граф из почти 1000 связей.
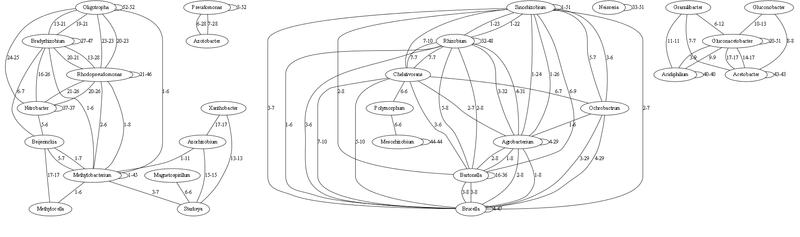
Здесь граф с наиболее сильными связями (опущены связи до 5 идентичных тРНК включительно)
Каждая связь характеризуется минимальным-максимальным числом совпадающих (идентичных на 100%) генов тРНК. Связь рода с самим собой означает число идентичных тРНК внутри этого рода (т.е. как различаются виды).
По сути это все надо еще визуально обработать, чтобы можно было бы объять визуально все это множество. На графе с 1000 связями есть много родов, которые не с кем не связаны — но если бы отобразить более слабые связи до 5 идентичных тРНК — то можно увидеть дальних родственников. (это я подумываю сделать следующим этапом, если есть желающие помочь — пишите).
По сути, на этом основании многое совпадает согласно текущей классификации. Число идентичных тРНК хорошо иллюстрирует дальность родов друг от друга, чем меньше идентичных тРНК — тем более древний предок. Те рода, которые имеют мало связей — наиболее древние (т.к. секвенируют сейчас, а их популяция в настоящие время представлена отдельными видами). Анализируя их можно построить достаточно точно процесс начальной эволюции.
upd. Убрал из графа двухсторонние связи (засоряли изображение). Общие число связей уменьшилось до 4551. Это позволило отобразить больший граф:
Скачать изображение можно тут (11.2 MB). Тут граф с наиболее сильными связями (опущены связи до 3 идентичных тРНК включительно)
Тогда видны связи (промежуточные виды) между двумя огромными доменами (выделяются на изображении, предположительно соответствуют Бета и Гамма протеобактериям), и другие детали. На сколько это соответствует текущей классификации надо сравнивать, но есть над чем подумать (просто детализация такова, что наверняка есть то, что не попало в текущую научную классификацию).
upd2 Используя yEd Graph Editor получилось отобразить полный граф. Ниже мини картинка.

Изображение получается плохо из-за связей не видно деталей, поэтому ниже файл в формате yEd Graph Editor, там по крайней мере можно увеличить, подвинуть и разглядеть. Если кто-то заинтересуется и сделает более обозримый граф — скажу спасибо :).
Граф «Систематика прокариот (505 родов и 4548 связей между ними)»
Вот теперь образовалась пауза, и я добил эту тему, и представляю результаты.
Метод
(кое-что повторю из предыдущей статьи, чтобы не заставлять новых читателей ее читать)
Основная критика статьи Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение» заключалась в следующей претензии "Нельзя рассматривать один ген в качестве мерила". С этим я полностью согласен, и этот эксперимент это исправляет в полной мере.
Немного цифр. Сейчас в NCBI есть порядка 2000 геномов бактерий (3723 локусов). При подготовке к эксперименту, я выделил все тРНК, которые помечены таким образом. Их оказалось более 40 тысяч уникальных вариаций. Но увы, среди них много ошибок (порядка 50%, см. предыдущие статьи, где это обсуждалось подробно).
Но я подумал, что можно пропустить этап полноценного исправления ошибок. Как это сделать? Я рассортировал указанные тРНК по длине и по наличию конца CCA на конце последовательности. Надо сказать, что последовательность CCA обязательна для любой тРНК, а длина может быть от 74 до 96 нуклеотидов.
В NCBI есть много чудес вплоть до тРНК из одного нуклеотида, или более 1300 :) (без улыбки не скажешь). Поэтому я убрал последовательности, которые имеют длину до 70 и больше 100, а также те которые не оканчиваются на CCA.
Их стало около 20000. Это наиболее вероятные тРНК, которые не содержат ошибок из NCBI. С оставшийся половиной тРНК — можно разобраться позже.
На самом деле, для планируемого эксперимента без разницы содержит ли ошибки данная конкретная последовательность длиной 70-100 нуклеотидов или нет. Почему? Так как я собираюсь перепроверить по геномам 2000 бактерий, действительно ли есть такие последовательности — ошибки будут исключены. А тРНК это на самом деле или нет это дело второе. Главное, что у разных организмов совпадают значительные участки ДНК. Совпадение последовательности длиной 70-100 в геномах — дело далеко не случайное.
Поэтому, что теперь я делаю. Беру этих 20000 тРНК и нахожу в каких бактериях они присутствуют. Если последовательность присутствует только в одном организме — это не интересно. И скорее всего это ошибочная последовательность. И таким образом отсеивается еще солидный процент ошибок.
Если же последовательность есть в более чем в одном организме — это одна ассоциация (связь) между двумя организмами.
Результаты
В первой статье был сделан важный вывод, что
Многовидовое происхождение сильно запутывает эволюционную картину, но с этим ничего не поделаешь — такова сложность видообразования, и нам нужно лишь их наиболее точно отразить в условиях, когда не все виды известны.
И поэтому для адекватного описания нужны не филогенетически деревья. Как минимум можно говорить о половых деревьях с двумя родителями (для усреднения), а в общем случае граф.
Также мне посоветовали отобразить граф с помощью Graphviz, что я и сделал. Но Graphviz зависает, когда число связей в графе больше 1000. А общий граф у меня получился на 6172 связи. Поэтому тут показываю лишь маленький фрагмент для наглядности. И даю ссылку на граф из почти 1000 связей.
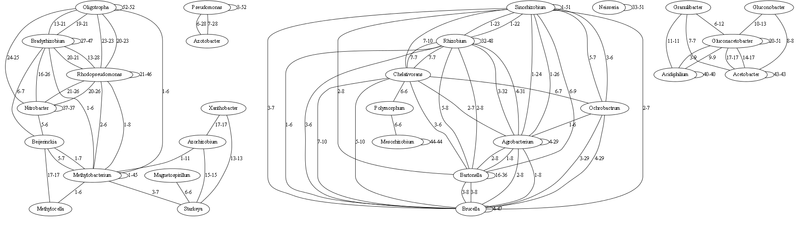
Здесь граф с наиболее сильными связями (опущены связи до 5 идентичных тРНК включительно)
Каждая связь характеризуется минимальным-максимальным числом совпадающих (идентичных на 100%) генов тРНК. Связь рода с самим собой означает число идентичных тРНК внутри этого рода (т.е. как различаются виды).
Некоторые выводы
По сути это все надо еще визуально обработать, чтобы можно было бы объять визуально все это множество. На графе с 1000 связями есть много родов, которые не с кем не связаны — но если бы отобразить более слабые связи до 5 идентичных тРНК — то можно увидеть дальних родственников. (это я подумываю сделать следующим этапом, если есть желающие помочь — пишите).
По сути, на этом основании многое совпадает согласно текущей классификации. Число идентичных тРНК хорошо иллюстрирует дальность родов друг от друга, чем меньше идентичных тРНК — тем более древний предок. Те рода, которые имеют мало связей — наиболее древние (т.к. секвенируют сейчас, а их популяция в настоящие время представлена отдельными видами). Анализируя их можно построить достаточно точно процесс начальной эволюции.
upd. Убрал из графа двухсторонние связи (засоряли изображение). Общие число связей уменьшилось до 4551. Это позволило отобразить больший граф:
Скачать изображение можно тут (11.2 MB). Тут граф с наиболее сильными связями (опущены связи до 3 идентичных тРНК включительно)
Тогда видны связи (промежуточные виды) между двумя огромными доменами (выделяются на изображении, предположительно соответствуют Бета и Гамма протеобактериям), и другие детали. На сколько это соответствует текущей классификации надо сравнивать, но есть над чем подумать (просто детализация такова, что наверняка есть то, что не попало в текущую научную классификацию).
upd2 Используя yEd Graph Editor получилось отобразить полный граф. Ниже мини картинка.

Изображение получается плохо из-за связей не видно деталей, поэтому ниже файл в формате yEd Graph Editor, там по крайней мере можно увеличить, подвинуть и разглядеть. Если кто-то заинтересуется и сделает более обозримый граф — скажу спасибо :).
Граф «Систематика прокариот (505 родов и 4548 связей между ними)»

{kind=link}
{kind=link}