Сразу надо сказать, что буду излагать вопрос о биовычислениях с определенной кибернетико-геометрической точки зрения. Это мое название и это направление не распространено. Уверен, что так будет легче понять тем кто не в теме этой биологической проблематики. Те кто уже в теме — готов и с вами подискутировать и показать почему традиционные методы не пригодны с точки зрения кибернетического подхода (но в этой статье не вы моя аудитория — уж извините, но уверен и вам она будет полезна как расширение мировоззрения на проблематику).
Практическое применение для биологов имеет больше вопрос сворачивания белков. В определенной степени очень много практических задач можно свести к этой задаче (знанию того как сворачивается белок), основная из которых — разработка лекарств по борьбе с вирусами и болезнями.
Но эта задача в общем виде не решена. Это как нерешенные задачи в математике, только с биологическим контекстом (см. парадокс Левинталя). Биологи могут лишь с определенной погрешностью увидеть путем биоэкспериментов состояние в уже свернутом состоянии, но проследить как это происходит пока не возможно. Но все это кроме того очень дорого. Почему и занимаются компьютерными вычислениями — это дешево, даже не смотря на то, что используется тысячи компьютеров в распределенных проектах.
Но введения хватит, далее с корабля на бал…
Во-первых, почему РНК, а не белок? Просто потому что это проще. Нельзя понять как сворачиваются белки, не понимая как сворачиваются более простые молекулы РНК. Мы ведь не биологи, и нам важен не практический биологический результат, а понимание процесса математически/кибернетически.
Вот есть такая игра FoldIt. И там же есть вопрос читателя:
Отвечаю. Эта игра крик отчаяния. Совершенно нет алгоритмов, которые сворачивают. Даже полный перебор не помогает. Парадокс Левинталя показывает, что расчет сворачивания молекулы может длится больше всей жизни Вселенной.
Игрокам предлагаю такой эксперимент в игре. Вначале вам дается молекула уже кое-как свернутая и с какой-то предопределенной структурой (спирали/листы/петли). Если вы сбросите и загрузите еще раз — вам дадут уже немного другую структуру и начальное состояние. Это начальное состояние для вас уже просчитывают специалисты и просто распределяют между вами (как между компами), что вам вычислять. Сделайте следующие. С помощью «резинок» растяните цепь белка в линию. Уберите из структуры все спирали и листы, т.е. превратите все в петлю (на самом деле просто цепь без структуры). До начала этих действий заметьте сколько вам давали баллов (оценка состояния). Сравните с тем что получилось. Видим разница не очень большая. Так ведь? Но теперь попробуйте свернуть хотя бы до того состояния которое было изначально. Ни один из имеющихся инструментов вам не поможет это сделать автоматически. Если вы не запомнили как примерно выглядит структура — у вас нет даже никаких гипотез как это сделать. Буду рад если как говорит Дэвид Бейкер
но я тут пессимист.
Во-вторых, биологи любят говорить о минимальной энергии свернутого белка, и якобы когда энергия минимальная — белок занимает наиболее стабильное состояние. Но это не доказано, это теория. На эту энергию влияет очень много факторов, которые рассчитать просто не возможно.
Но мы поступим совершенно по другому. Мы будем учитывать ТОЛЬКО важность образования водородных связей и отсутствие запрещенных ковалентных (что это такое объясню позже). Почему? Потому, что в моих экспериментах этого оказалось во многом достаточно, чтобы получить результаты сопоставимые с результатами серьезных исследовательских проектов (не количественно (т.к. у меня всего один компьютер), а качественно).
И главное, постулируем этот основной принцип, кибернетико-геометрического подхода:
Теперь несколько картинок (кстати как регулировать размер картинки подскажите кто знает: стоит height=«20» width=«20» — но как видим размер большой):
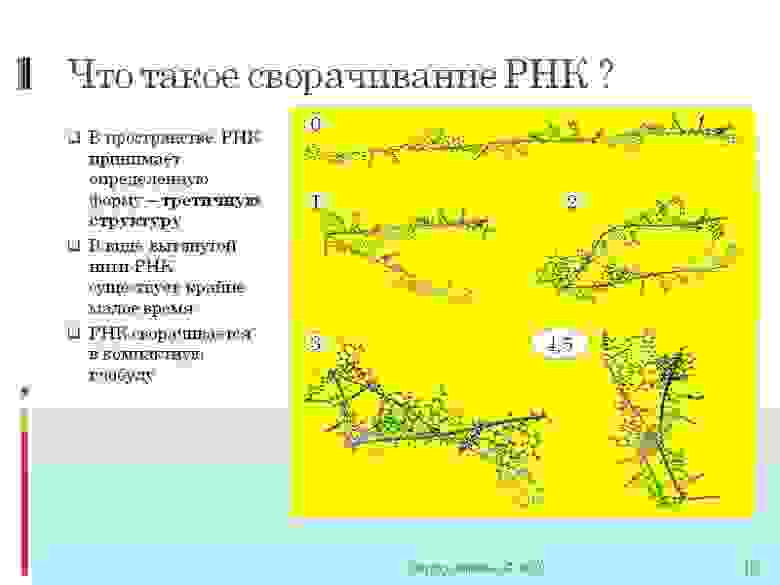
На картинке: шаг 0. Видим цепь РНК. Кто читал мою программно направленную статью — это как раз и есть начальный граф цепи о котором идет там речь.
Дальше вы видите как цепь за несколько шагов стремится свернуться в спираль. Хорошо построенная спираль на следующем рисунке

Итак, объясняю что такое ковалентные и водородные связи и на первый раз достаточно.
В шаге 0 (первый рисунок) — вы видите, что точки соединены линиями. Точки это отдельные атомы. А линии — это разрешенные ковалентные связи. В процессе сворачивания нельзя допустить, чтобы образовались какие-то еще ковалентные связи, грубо говоря, чтобы разрешенные ковалентные связи пересекались бы. В игре FoldIt это как раз показывается красным «шиповником» и сильно штрафуется очками. Просто в реальности такие связи образоваться не могут — мало для этого энергии, а программно конечно легко так накрутить что легко столкнуть. Это и есть запрещенные ковалентные связи.
Водородные связи видны на втором рисунке красные прерывистые линии между атомами. Эти связи более легко образуются и в основном то они и удерживают РНК в свернутом состоянии.
Во второй части мы поговорим какие бывают водородные связи, как их образование описать математически и начальные шаги к тому по какой логике начинать сворачивать РНК. Но хотелось бы обратной связи — пишите, кто что понял, и готовы ли читать вторую часть. Или я чего-то упустил для понимания, дальше будет только сложнее, поэтому лучше говорите сразу.
Практическое применение для биологов имеет больше вопрос сворачивания белков. В определенной степени очень много практических задач можно свести к этой задаче (знанию того как сворачивается белок), основная из которых — разработка лекарств по борьбе с вирусами и болезнями.
Но эта задача в общем виде не решена. Это как нерешенные задачи в математике, только с биологическим контекстом (см. парадокс Левинталя). Биологи могут лишь с определенной погрешностью увидеть путем биоэкспериментов состояние в уже свернутом состоянии, но проследить как это происходит пока не возможно. Но все это кроме того очень дорого. Почему и занимаются компьютерными вычислениями — это дешево, даже не смотря на то, что используется тысячи компьютеров в распределенных проектах.
Но введения хватит, далее с корабля на бал…
Во-первых, почему РНК, а не белок? Просто потому что это проще. Нельзя понять как сворачиваются белки, не понимая как сворачиваются более простые молекулы РНК. Мы ведь не биологи, и нам важен не практический биологический результат, а понимание процесса математически/кибернетически.
Вот есть такая игра FoldIt. И там же есть вопрос читателя:
не совсем понял как ручной перебор сокращает моделирование на порядок. То как люди руками свернут белок тоже в общем то случайно, люди ведь даже приблизительно не понимают как его надо сворачивать. А если считать что люди просто отбросят ряд нереальных вариантов по какому либо признаку, почему нельзя это алгоритмизировать в folding@home?
Отвечаю. Эта игра крик отчаяния. Совершенно нет алгоритмов, которые сворачивают. Даже полный перебор не помогает. Парадокс Левинталя показывает, что расчет сворачивания молекулы может длится больше всей жизни Вселенной.
Игрокам предлагаю такой эксперимент в игре. Вначале вам дается молекула уже кое-как свернутая и с какой-то предопределенной структурой (спирали/листы/петли). Если вы сбросите и загрузите еще раз — вам дадут уже немного другую структуру и начальное состояние. Это начальное состояние для вас уже просчитывают специалисты и просто распределяют между вами (как между компами), что вам вычислять. Сделайте следующие. С помощью «резинок» растяните цепь белка в линию. Уберите из структуры все спирали и листы, т.е. превратите все в петлю (на самом деле просто цепь без структуры). До начала этих действий заметьте сколько вам давали баллов (оценка состояния). Сравните с тем что получилось. Видим разница не очень большая. Так ведь? Но теперь попробуйте свернуть хотя бы до того состояния которое было изначально. Ни один из имеющихся инструментов вам не поможет это сделать автоматически. Если вы не запомнили как примерно выглядит структура — у вас нет даже никаких гипотез как это сделать. Буду рад если как говорит Дэвид Бейкер
искренне верит, что где-то в мире живут таланты, у которых есть врождённая способность просчитывать в уме 3D-модели протеинов. Какой-нибудь 12-летний мальчик из Индонезии увидит игру и сможет решить задачи, которые не под силу даже суперкомпьютеру
но я тут пессимист.
Во-вторых, биологи любят говорить о минимальной энергии свернутого белка, и якобы когда энергия минимальная — белок занимает наиболее стабильное состояние. Но это не доказано, это теория. На эту энергию влияет очень много факторов, которые рассчитать просто не возможно.
Но мы поступим совершенно по другому. Мы будем учитывать ТОЛЬКО важность образования водородных связей и отсутствие запрещенных ковалентных (что это такое объясню позже). Почему? Потому, что в моих экспериментах этого оказалось во многом достаточно, чтобы получить результаты сопоставимые с результатами серьезных исследовательских проектов (не количественно (т.к. у меня всего один компьютер), а качественно).
И главное, постулируем этот основной принцип, кибернетико-геометрического подхода:
мы идеализируем процесс сворачивания, не рассматривая никакие другие взаимодействия, кроме водородных связей. Таким образом, в моделировании мы намеренно исходим из упрощения, идеализации, как бы отвечая на вопросы: «как пойдет ход сворачивания, если РНК будет стремиться только к образованию водородных связей?» и «каков “чистый” вклад образования водородных связей в процесс сворачивания?» (это из моей научной статьи)
Теперь несколько картинок (кстати как регулировать размер картинки подскажите кто знает: стоит height=«20» width=«20» — но как видим размер большой):
На картинке: шаг 0. Видим цепь РНК. Кто читал мою программно направленную статью — это как раз и есть начальный граф цепи о котором идет там речь.
Дальше вы видите как цепь за несколько шагов стремится свернуться в спираль. Хорошо построенная спираль на следующем рисунке

Итак, объясняю что такое ковалентные и водородные связи и на первый раз достаточно.
В шаге 0 (первый рисунок) — вы видите, что точки соединены линиями. Точки это отдельные атомы. А линии — это разрешенные ковалентные связи. В процессе сворачивания нельзя допустить, чтобы образовались какие-то еще ковалентные связи, грубо говоря, чтобы разрешенные ковалентные связи пересекались бы. В игре FoldIt это как раз показывается красным «шиповником» и сильно штрафуется очками. Просто в реальности такие связи образоваться не могут — мало для этого энергии, а программно конечно легко так накрутить что легко столкнуть. Это и есть запрещенные ковалентные связи.
Водородные связи видны на втором рисунке красные прерывистые линии между атомами. Эти связи более легко образуются и в основном то они и удерживают РНК в свернутом состоянии.
Во второй части мы поговорим какие бывают водородные связи, как их образование описать математически и начальные шаги к тому по какой логике начинать сворачивать РНК. Но хотелось бы обратной связи — пишите, кто что понял, и готовы ли читать вторую часть. Или я чего-то упустил для понимания, дальше будет только сложнее, поэтому лучше говорите сразу.
