Comments 33
А поиграться дадите?
В принципе без проблем, если есть желание покопаться в исходниках.
Дело в том, что пока я задаю положения частиц тупо в программном коде. В некоторых случаях это удобно — например «темную» поверхность я задавал используя модуль косинуса (чтобы сделать зубчики).
Дело в том, что пока я задаю положения частиц тупо в программном коде. В некоторых случаях это удобно — например «темную» поверхность я задавал используя модуль косинуса (чтобы сделать зубчики).
Есть куча доступных кодов как для MD, так и для много чего другого типа DFT и так далее. Есть MD, специализированные под разные вещи. Я например, в свое время слегко модифицировал под свои цели (смотрел, как ведет себя углеродная нанотрубка под давлением) код Бреннера: www.eng.fsu.edu/~dommelen/research/nano/brenner/
Большая часть различных кодов написана на Фортране, но есть и на C.
вот пример того, что получилось у меня:
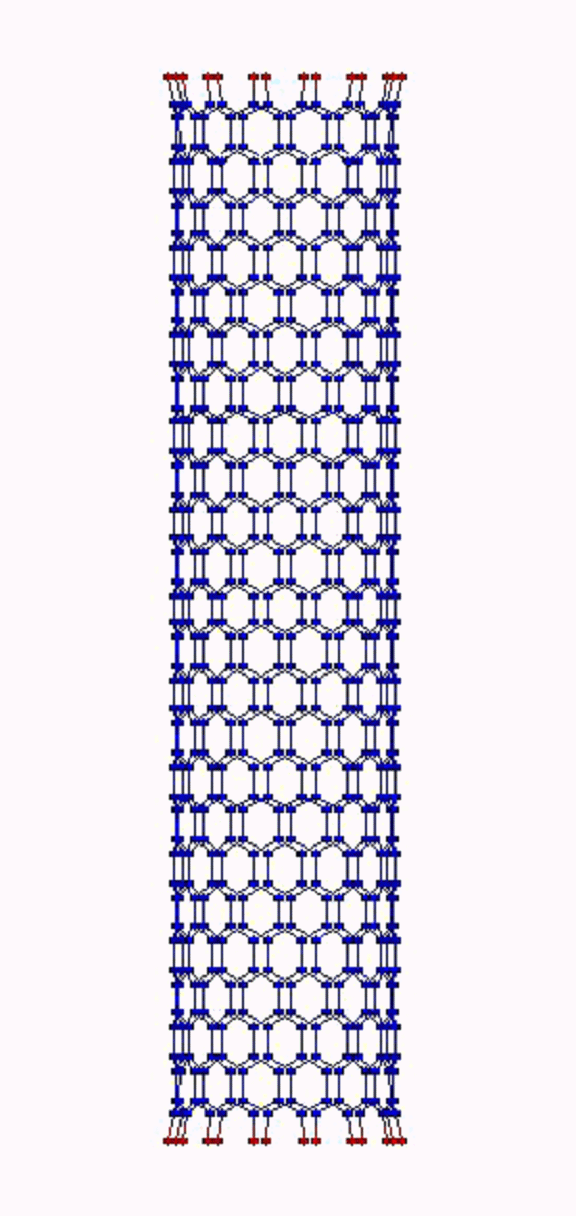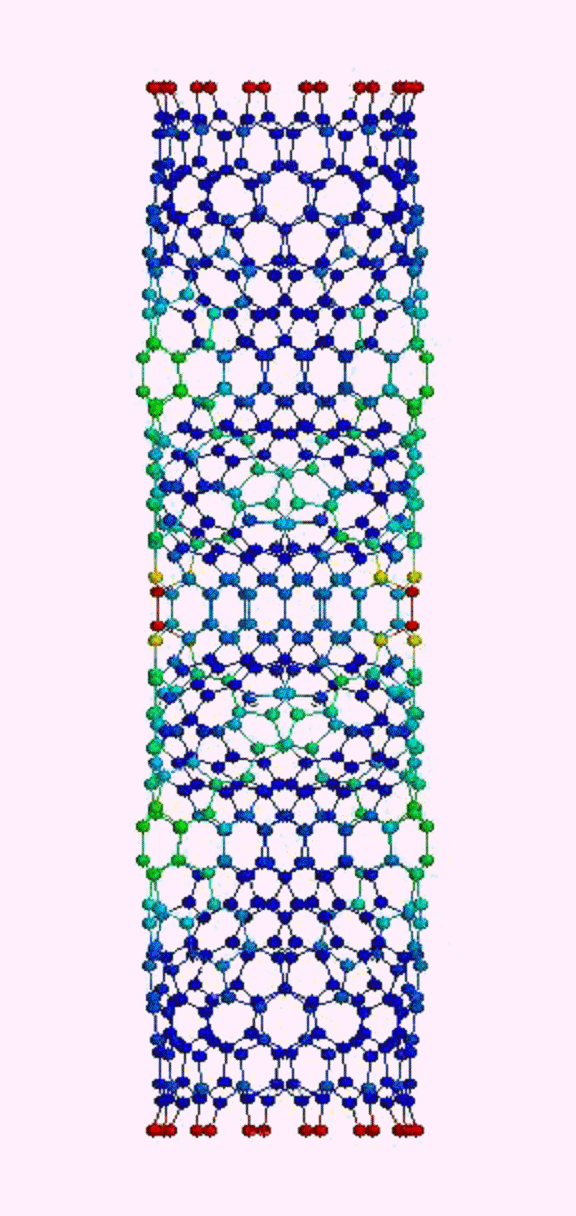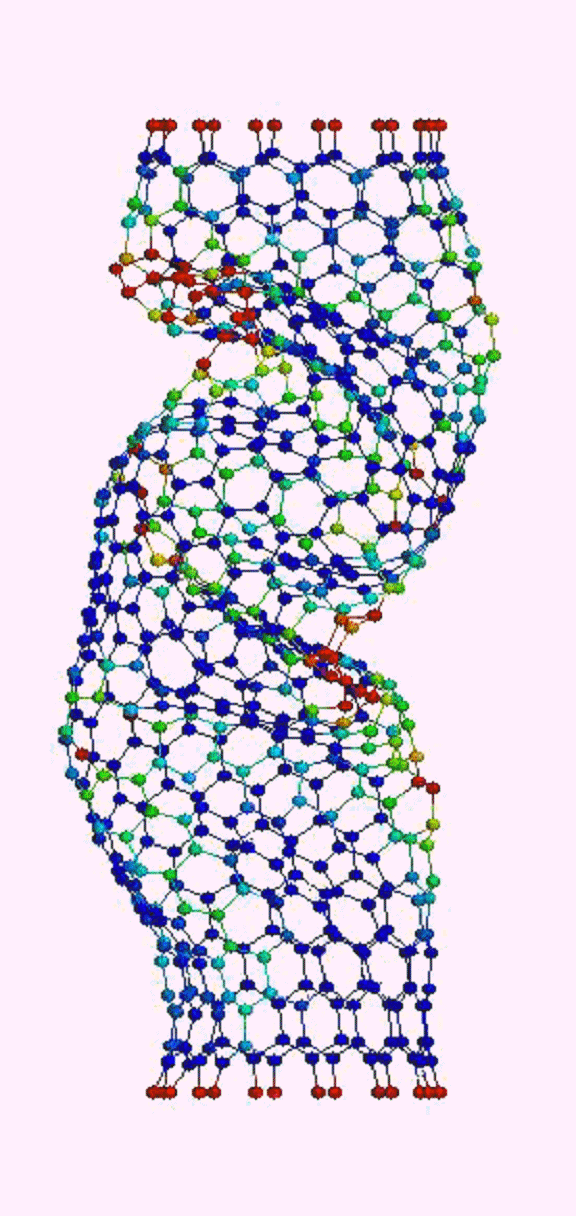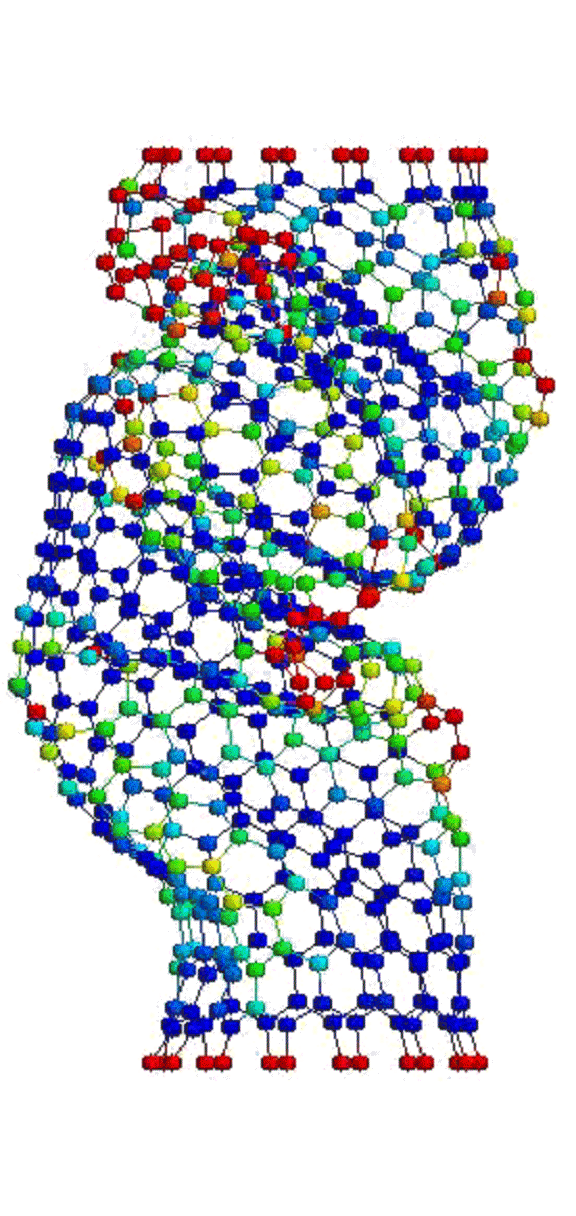
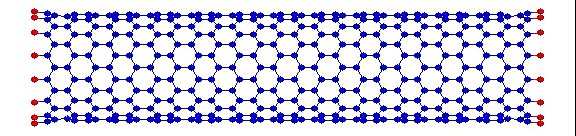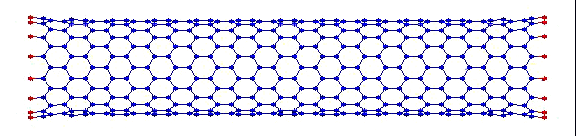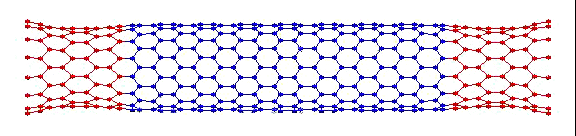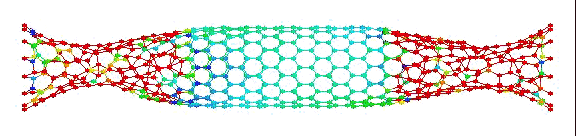

Это, соответственно, компрессия, растяжение, и сгинание (извините, если не совсем по-русски, короче, compression, tension and bending)…
Делалось это кстати все в рамках курса Computational Physics.
Большая часть различных кодов написана на Фортране, но есть и на C.
вот пример того, что получилось у меня:
Это, соответственно, компрессия, растяжение, и сгинание (извините, если не совсем по-русски, короче, compression, tension and bending)…
Делалось это кстати все в рамках курса Computational Physics.
Упс, сорри за первую картинку, она не должна быть такой большой.
Оптимизировал для Ваc картинки, получилось примерно вдвоее легче. Подмените, пожалуйста, на хостинге исходные файлы теми, что лежат по адресу — читать народу будет легче.
ifolder.ru/21199625
ifolder.ru/21199625
Сорри, сорри, сорри, картинки не у меня лежат, а у преподавательницы по курсу, я хотлинкнул, я ей сдавал все в виде видео, я так понял, они гифки сделала. Насчет визуального размера — у нее смотрелся нормально, насчет веса — я даже и не ожидал, что можно 10 мегов таким образом набрать. Удалить комментарий тоже не могу, так что сорри, сорри, сорри…
А как насчет задачи смоделировать формирование солнца и планет из хаотичной звездной пыли? Берем N-частиц, задаем гравитационное взаимодействие, запускаем и смотрим смогут ли они из хаотичного газа слипнуться в сгусток солнца и планеты вокруг и как будут двигаться?
Памяти не хватит. Даже если для каждого атома потребуется 1 байт, то чтобы представить 1 моль вещества потребуется 6*10^23/10^9 = 6*10^14 Гб памяти.
Я только о моделировании макроразмеров мечтаю, а вы уже солнечные системы!
Интересная задача для такого метода, но представте какое же количество частиц газа необходимо будет для моделирования целой системы!
Если же мы возьмем абстрактый «булыжник» размером в несколько км как частицу(чтобы сделать реальным время расчета) — тут потребуется уже совсем другой потенциал. Его еще надо придумать.
Интересная задача для такого метода, но представте какое же количество частиц газа необходимо будет для моделирования целой системы!
Если же мы возьмем абстрактый «булыжник» размером в несколько км как частицу(чтобы сделать реальным время расчета) — тут потребуется уже совсем другой потенциал. Его еще надо придумать.
Это лучше стандартными методами гидродинамики обсчитывать. Уравнения Навье-Стокса, метод конечных элементов, и тп. Другой уровень абстракции, так сказать.
Если всё это на молекулярном уровне обсчитать, надо бы будет потом приглядеться — не получилось ли на поверхностях некоторых планет тонкой плёнки молекул, скомбинировавшихся в жизнь и цивилизации… %)
Немного в другом масштабе, но на кафедре ТеорМеха СПбПУ это уже сделано: Земля-Луна.
В простейшем варианте MD, действительно, хорошо ускоряется на GPU, но только до тех пор пока на арену не выходят дальнодействующие взаимодействия, например, электростатические. Корректный учет зарядов в системах приближенных к реальным — это огромная головная боль и существующие алгоритмы, позволяющие делать это не так уж и хорошо параллелятся на той же CUDA.
А вообще в плане практического моделирования такого рода систем я бы посоветовал посмотреть на пакет ESPResSo. Он многофункциональный, но если пошаманить опциями при сборке и отключить все ненужное, то можно получить довольно шустрое создание, параллельное из коробки (через MPI). У него есть только один минус: все параметры, сам цикл моделирования и обработки надо будет писать на Tcl…
А вообще в плане практического моделирования такого рода систем я бы посоветовал посмотреть на пакет ESPResSo. Он многофункциональный, но если пошаманить опциями при сборке и отключить все ненужное, то можно получить довольно шустрое создание, параллельное из коробки (через MPI). У него есть только один минус: все параметры, сам цикл моделирования и обработки надо будет писать на Tcl…
Автор, потенциал равен интегральчику от f®, ну или сила равна производной от f®, а не тупо 1/r * f® ;). Ну и да, ИМХО, можно было бы использовать существующие решения для МД, например LAMMPS, а писать самому только визуализацию :). Потому что с интегрированием уравнений движения не всё так просто, надо использовать правильные схемы, которые являются хотя бы консервативными, а не просто Эйлера (или кого-нибудь другого :)). А в остальном всё замечательно :)
Хабрапарсеру привет… f ( r ), а не f®:
А вообще, формула немного странная у Вас используется, ИМХО
Всё же вроде как очень просто —

И не надо никаких дополнительных и сбивающих с толку формул :)
Всё же вроде как очень просто —

И не надо никаких дополнительных и сбивающих с толку формул :)
… диссипативную составляющую ...
Давно меня так не торкало…
У меня диплом по молекулярной динамике был. В моей проге можно создавать различные решетки, задавать потенциалы и делать другие штуки. Могу выложить.
ifolder.ru/21204653 — вот такая вот программа. На ней я тоже всякие исследования делал. Тоже статью думал написать, но было лень.
А вы не задумывались насчет GROMACS?
Кстати, по молекулярной динамике есть хорошая книжка Computer simulation of liquids (и <a href=«www.filestube.com/abb88c84ad44045503ea,g/Computer-Simulation-of-Liquids.html»бесплатно). Она 1989 года, но нам ее рекомендовал в универе бывший препод, который сейчас профессионально занимается MD в Германии. Осторожно: ОЧЕНЬ много математики (даже кватернионы есть:-)).
Кстати, молекулярная динамика--это не только расчет траектории движения одной системы в фазовом пространстве, но и расчет распределения систем по состояниям фазового пространства при заданных внешних условиях. И там совсем другие методы.
Кстати, по молекулярной динамике есть хорошая книжка Computer simulation of liquids (и <a href=«www.filestube.com/abb88c84ad44045503ea,g/Computer-Simulation-of-Liquids.html»бесплатно). Она 1989 года, но нам ее рекомендовал в универе бывший препод, который сейчас профессионально занимается MD в Германии. Осторожно: ОЧЕНЬ много математики (даже кватернионы есть:-)).
Кстати, молекулярная динамика--это не только расчет траектории движения одной системы в фазовом пространстве, но и расчет распределения систем по состояниям фазового пространства при заданных внешних условиях. И там совсем другие методы.
интересная статья, спасибо
Sign up to leave a comment.
Программирование молекулярной динамики